18 звезд, которые воспитывают особенных детей
Содержание:
Прогноз
Прогноз течения заболевания неоднозначен, некоторые дети с течением времени все же получают минимально необходимый набор навыков и умений, постепенно учатся заботиться о себе и общаться с другими людьми, у других детей такие навыки не вырабатываются в течении всей их жизни.
Все зависит от степени тяжести генетических нарушений в строении 15 хромосомы, адекватности и регулярности терапевтических и коррекционных процедур, отношения родителей к больному ребенку.
Синдром Ангельмана – редкое заболевание, обусловленное наличием определенных нарушений и генетических отклонений в строении 15 хромосомы. Заболевание может передаваться по наследству, а может развиваться спонтанно.
Тем не менее, наличие генетических мутаций у родителей существенно повышает риск развития недуга.
Ребенок, страдающий данной патологией имеет отставание в физическом и психическом развитии, и эти факторы практически нельзя устранить.
Заболевание невозможно полностью вылечить, однако, соблюдение рекомендаций врача позволит ребенку приобрести хотя бы минимальный набор жизненно-необходимых навыков и умений.
О болезни Ангельмана вы можете узнать из этого видео:
Формы
Синдром Ангельмана связан с четырьмя вариантами генетических мутаций:
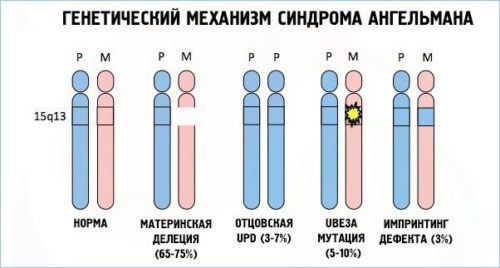
- Вновь возникшей хромосомной мутацией, которая связана с потерей участка хромосомы в локусе 15 q11 — q13 . Данная мутация – причина около 80 % всех случаев заболевания.
- Одноотцовской дисомией, которая связана с потерей материнского локуса (отсутствие генетического материала матери). Данный вариант встречается редко (около 5% всех случаев).
- Дефектом ряда генов, подверженных геномному импринтингу (ГИ). Данные дефекты возникают у 2-4% больных в результате непосредственного нарушения импринтинга (различия в преобразовании информации гена в белок или РНК, которые зависят от происхождения гена). Чаще всего возникает в результате утраты центра регуляции ГИ. Дефекты ГИ без утраты центра регуляции являются результатом спонтанной мутации, повторение которой – большая редкость.
- Спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A. Данный ген кодирует деятельность убиквитинлигазы (фермент, участвующий в сложном процессе распада белков). Дефицит данного фермента относится к молекулярным механизмам синдрома.
Установить форму заболевания у 7-9 % в настоящее время не представляется возможным.
Симптомы
Первые признаки синдрома Ангельмана появляются к концу первого года жизни ребенка. Более явными симптомы становятся по достижению двухлетнего возраста. Синдром характеризуется полиморфностью клинических проявлений. Больных можно узнать сразу. Такие дети имеют характерный внешний вид.

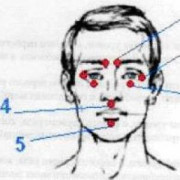
- Особенности внешнего вида больного ребенка: несоответствие маленькой головы и нормального туловища, большой рот, постоянная улыбка, широкие межзубные промежутки, узкие губы, высунутый широкий язык, неправильный прикус, плоский затылок, гладкие ладони, заостренный подбородок, гипопигментированная кожа, стробизм, сколиоз, ходьба на прямых ногах.
- Нарушение психоэмоционального развития проявляется отставанием в умственном развитии, избыточной суетливостью, дружелюбностью, беспричинным смехом, эйфорией.
- Неврологические симптомы: тремор конечностей, атаксия, дискоординация движений, потеря равновесия, гипотония мышц, расстройство сна, истерия, нарушение речи, гиперактивность и гипервозбудимость, сложности с обучением.
- Двигательные расстройства: слабый контроль за движением языка и его беспричинное высовывание, трудности с глотанием и сосанием, поднятые или согнутые во время ходьбы руки, частое слюнотечение, неуемная жажда, излишне активные жевательные движения, нарушение мелкой моторики, эпилепсия.
Дети с синдромом Ангельмана склонны к невербальному общению: воспринимают устную речь, но не могут высказать свои мысли. Они практически не участвуют в разговоре, поскольку их словарный запас состоит из 10-20 слов, необходимых в быту. У больных присутствуют стереотипии – повторяющиеся взмахи руками, кручения кистями и частое хлопанье в ладоши, а также прочие действия, не характерные для здоровых людей. Шаткая походка сопровождается резкими подергиваниями руками, что делает человека похожим на марионетку. Больные чувствуют себя комфортнее в воде и плохо переносят душные помещения и высокую температуру.
С возрастом клиническая картина болезни значительно изменяется. Судороги и эпиприступы возникают все реже или исчезают совсем. Пациенты становятся более спокойными, у них налаживается сон. Мужчины и женщины с данным синдромом имеют моложавую внешность, скрывающую их истинный возраст. Половое созревание у них происходит вовремя, возможно даже рождение детей.

К симптомам патологии у взрослых относятся нарушения мелкой моторики и недержание мочи. Больные не могут справиться с пуговицами и молниями на одежде, но при этом спокойно пользуются вилкой и ложкой. Они дружелюбные, любящие и ласковые. Ожирение – распространенное отклонение среди взрослых больных, особенно женщин. Люди с синдромом Ангельмана склонны к расстройствам вегетативного и соматического характера. Они плохо переносят жаркую погоду, часто страдают от запоров, пищеводного рефлюкса.
Поскольку заболевание является врожденным, все перечисленные аномалии могут проявиться сразу после родов. Современные методы диагностики позволяют выявить синдром внутриутробно и предотвратить рождение больного ребенка.
Лечение
Синдром Ангельмана является врожденной генетической аномалией; в настоящее время специфические способы его лечения не разработаны, однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. Младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии).
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0.3 мг мелатонина за 30 минут-1 час перед сном улучшает сон пациентов с синдромом Ангельмана. Нарушения стула регулируются назначением легких слабительных.
Приступы лечатся так же, как эпилепсия. Дети с синдромом Ангельмана часто испытывают больше одного типа приступов. Показана электроэнцефалография.
Нежелательное поведение. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложность в общении с другими людьми. Врачи США показывают, что для аутичных детей внутривенные инъекции гормона секретин (найденного в поджелудочной железе) успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана.
Генетические мутации повышают риск появления заболевания
Синдром Ангельмана встречается нечасто — примерно 1 случай на 10 – 20 тыс. новорожденных малышей. Основной причиной патологии является потеря в 15 хромосоме копий нормальных материнских генов, что происходит по причине нарушения деления данной хромосомы. Кроме того заболевание может возникнуть в связи с мутацией отцовских генов, отцовской дисомией или трисомией.
В норме здоровый человек получает от матери и отца по одной копии 15 хромосомы. Если ребенок получил от кого-то из родителей генетически измененную копию (особенно от матери, так как у нее копии генов более сильные, чем у отца), у ребенка развивается синдром Ангельмана.
Заболевание носит имя Гарри Ангельмана, британского врача-педиатра, который впервые дифференцировал данный синдром в 1965. Тогда он получил название синдрома счастливой марионетки, однако сегодня этот термин не используется, так как это было признано пренебрежительным.
Доктор Ангельман занимался лечением нескольких детей со сходными симптомами и предполагал наличие общего диагноза. Доказать диагноз и получить точные данные на тот момент было не возможно, ввиду отсутствия технологий, которые доступны сегодня. Свои догадки врач отразил в статье под названием «Дети марионетки».
Появление синдрома Ангельмана связано с наличием у родителей будущего ребенка различных хромосомных аномалий. Среди таких отклонений обычно называют:
- трисомию хромосом – присутствие одной или нескольких лишних хромосом в хромосомном наборе;
- инверсию – разворот одного из участков хромосомы на 180 градусов, при этом часть хромосомы пропущена, а гены располагаются в противоположном порядке;
- микроделецию, которая является результатом перестройки Y-хромосомы и обмена участками между хромосомами, наблюдается небольшое количество хромосом, а также может отсутствовать один из генов;
- делецию – нехватку одного из участков хромосомы;
- транслокацию – перенос или присоединение участка одной хромосомы к другой хромосоме;
- дупликацию – копирование части хромосом, результатом чего становится лишний генетический материал;
- кольцевую хромосому – на концах хромосомы отсутствует генетический материал, при этом новообразованные концы соединяются в виде кольца.
Генные мутации, которые могут вызвать развитие синдрома
Рассматриваемая болезнь обязана своим названием врачу-педиатру Гарри Ангельману, который впервые диагностировал отклонение в 1965 году и назвал пациентов кукольными детьми.
Именно картина «Мальчик-марионетка» подарила специалисту уверенность в собственной правоте. Картина отображает смеющегося мальчика, который очень напомнил Гарри его пациентов и он решает написать о них общую статью, которая в дальнейшем получила название «Дети-марионетки».
Признаки синдрома Ангельмана
В 1965 году издается научная работа, интерес о которой был забыт до восьмидесятых, когда упоминания о патологии появились в медицинской среде. Закономерность отсутствия части 15-й хромосомы была установлена только в 1987 году. Поскольку диагноз «синдром счастливой марионетки» унижал и пугал многих родителей, было решено использовать фамилию Ангельмана. Среди современных ученых, изучающих данное явление, стоит выделить М. Б. Миронова и К. Ю. Мухина.
В качестве факторов риска и провоцирующих элементов стоит рассмотреть родительские хромосомные аномалии.
- Кольцевая хромосома – удаление генетического материала с концов хромосомы и соединение новообразованных концов в кольцо.
- Дупликация — частичное повторение хромосом и соответствующее появление лишнего генетического материала.
- Транслокация – присоединение к хромосоме элемента другой хромосомы.
- Делеция предполагает отсутствие одного из секторов хромосомы.
- Микроделеция является следствием перестройки Y-хромосомы и обмена секторов между половыми хромосомами при мейозе. Немного хромосом при этом отсутствует, чаще всего одного из генов тоже нет.
- Инверсия – разворот на 180 градусов одного из участков хромосомы, порядок расположения генов обратный, а часть хромосомы опущена.
- Трисомии хромосом обусловлена наличием одной или больше лишних хромосом в наборе. Причиной такого генетического дефекта является нерасхождение хромосом при делении.
Симптомы
Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно нарастает задержка развития, ранее освоенные навыки сохраняются, но приобретение новых происходит медленно. Дети умеют сидеть, ползать, брать предметы и перекладывать их из руки в руку, поддерживать визуальный контакт, гулить и лепетать. Ходьба дается с трудом, нарушено чувство равновесия, наблюдаются частые падения, ушибы о мебель. Выявляется тремор и хаотичные движения конечностями, особенно руками. Речевые расстройства представлены как задержками, так и полным отсутствием экспрессивной речи. Дети либо совсем не говорят, либо используют лепет, простые слоги и слова общим объемом не более 10 единиц. Сохраняется понимание обращенной речи, стремление к общению, использование невербальных средств коммуникации: жестов, мимики, опосредованных знаков.
Основное поведенческое нарушение – гиперактивность. Дети часто веселятся и смеются без объективной причины, двигательно расторможены, неусидчивы, нецеленаправленны. У многих возникает патологическая привязанность к определенной игрушке или предмету быта, при появлении которого настроение сразу повышается, капризность и плач сменяются смехом. Концентрация внимания снижена, переключаемость быстрая и ненаправленная. Имеются трудности обучения, стойкое снижение интеллектуальных функций. Легко закрепляются стереотипии: раскачивание тела, размахивание руками. У 80% пациентов отмечается микроцефалия, недостаточный объем черепной коробки, эпилептическая активность мозга. Редко наблюдается снижение контроля движений языка, которое проявляется трудностями сосания груди или соски, последующим недостатком массы тела.
Характерные особенности внешности детей – косоглазие, сколиотическое искривление позвоночника, увеличение зубов и губ, разряжение зубного ряда, уплощение затылка, выступание вперед подбородка. Язык часто высунут, рот приоткрыт в улыбке. Развивается мышечная дистония, выраженность рефлексов сухожилий повышается, формируя специфичность моторики: пациенты ходят на прямых несгибающихся ногах, плечи приподнимают, руки сгибают в локтях. Своеобразный симптом – тяга к воде. Большинство детей чувствуют себя спокойнее в водной среде, им нравится плескаться в ванной, играть с корабликами в тазу.
Каким будет ребенок с синдромом куклы?
Возможно, вы заметили, что ваш малыш имеет задержку в развитии и обратились за помощью к педиатру. Из-за того, что это редкое заболевание, врачи обычно не подозревают синдром Ангельмана у детей.
Когда родителям говорят, диагноз – синдром смеющейся куклы, они не знакомы с характеристиками этого расстройства. Например, судороги, проблемы со сном, отклонения в поведении, смех, невербальное общение, тяжелые когнитивные нарушения, задержка в развитии речи.
Ранний диагноз полезен. Во-первых, у вас больше времени, чтобы узнать о том, что значит воспитывать ребенка с особыми потребностями. Это может быть пугающим, поскольку есть много факторов, которые следует учитывать, это: специальное образование; врачи и узкие специалисты.
Диагностика также означает раннее начало терапии и доступ к услугам, которые, имеют огромную пользу для малыша.

Не менее важна и поздняя диагностика. Хотя родители / опекуны могут подозревать, что их ребенок имеет особые потребности, получение генетического диагноза открывает двери для широкой базы знаний и стратегий, являющиеся эффективными в АС, а также для доступа к сообществу людей имеющим сходный опыт.
Частые вопросы
Как будет выглядеть ребенок? Это естественный вопрос – сможет ли он сидеть или ходить. Вы можете спросить: «Будет ли ребенок говорить или использовать язык жестов? Как будет играть с другими детьми? Пойдет ли в школу? Сможет ездить на велосипеде? Как будет развиваться? »
Хотя никто не может предсказать это точно, мы знаем, что не все дети с AS испытывают одинаковую степень и тяжесть симптомов. Ваш ребенок является одновременно продуктом ~ 29 000 нормально функционирующих генов.

Ошибочно предполагать, что все характеристики, будут присутствовать у каждого человека до степени, описанной клиницистами для синдрома в целом.
Оценивая достижения в области развития или прогресс в таких областях, как «когда ребенок начал сидеть, ходить, играть в игры». Разница велика даже среди детей с аналогичным генотипом.
Особенности питания
Человеку с синдромом Ангельмана требуется питательная диета. Учитывая проблемы с кормлением и рефлюксом, полезно обратиться за советом к диетологам. Если ребенок не может принимать твердую пищу, родители должны проконсультироваться с медицинским работником о G-образной трубке, обеспечивающей питательные вещества через хирургическое отверстие в желудке.
Заключительная мысль, состоит в том, что, хотя человек рождается с потерей функции одного гена, UBE3A (ген «Angelman»), существуют тысячи других полностью функциональных. Да, диагноз станет изменением жизни, но многие другие факторы влияют на развитие и определяют ее общее качество.
Лучшее, что может сделать любой родитель, – это получить информацию. Найдите отличного врача. Ищите других родителей и присоединяйтесь к организации синдрома Ангельмана, которая наилучшим образом соответствует вашим ценностям.
Мелкие чудеса происходят каждый день. Оставляйте дверь открытой, чтобы впустить их.
(Все люди, изображенные в этой статье, имеют синдром Ангельмана).
Съёмочная группа
- Автор сценария: Алёна Алова по роману Дины Рубиной «Синдром Петрушки»
- Режиссёр-постановщик: Елена Хазанова
- Оператор-постановщик: Азиз Жамбакиев
- Художник-постановщик: Наталья Навоенко
- Композитор: Николя Рабеюс
- Санкт-Петербургский симфонический оркестр
- Хореограф: Раду Поклитару
- Директор картины: Надежда Попова
- Со-продюсеры: Ундине Филтер, Томас Крал, Пьер-Андре Тьебо совместно с Антонио Эксакустос, Йозеф Райдингер
- Продюсеры: Илья Гаврилов, Александр Новин, Дмитрий Аронин, Ася Темникова, Елена Бренькова, Анна Качко
- Художественный руководитель: Евгений Миронов
Симптомы синдрома Ангельмана
Проявления расстройства в психо-эмоциональной сфере:
- задержка развития, в том числе отсутствие лепета и ползания в период от 6 месяцев до 1 года;
- угнетенная обучаемость;
- отсутствие речи либо минимальный навык говорения;
- немотивированный частый смех/улыбка;
- тип личности можно описать как радостный, экзальтированный.
Симптомы расстройства на соматическом уровне:
- приступы эпилепсии, которые обычно возникают в возрасте от 2 до 3 лет;
- затрудненная ходьба/перемещение, скованные или резкие угловатые движения из-за неспособности контролировать произвольные движения (атаксия);
- необычное поведение, такое как внезапные взмахи рук, ходьба с приподнятыми руками и/или на негнущихся ногах;
- небольшой размер головы с уплощением в затылочной зоне (микробрахицефалия);
- волосы, кожа и глаза светлого оттенка (гипопигментация);
- иногда у детей наблюдаются широкий рот, редкие зубы и высунутый язык. В большинстве случаев черты лица пациентов не отличаются чем-то необычным.
Отдельно следует остановиться на особенностях речевого развития и моторики, характерных для людей с синдромом Ангельмана.
У большинства детей с этим расстройством отмечают крайне скудный словарный запас, который может быть ограничен несколькими словами (до одного-двух десятков). Это обусловливает значительные трудности социализации ребенка, у которого наблюдается синдром Петрушки.
При этом дети, имеющие такое расстройство – как правило – понимают не только простые команды, но и способны усваивать информацию в объеме, который значительно превышает их возможности общения и выражения мыслей.
Что касается моторики, то кроме описанных выше проявлений, дети-«Петрушки» могут часто держать руки приподнятыми и согнутыми в локтях и запястьях, и периодически взмахивать руками в моменты возбуждения или во время ходьбы. Именно эта схожесть с движениями марионетки в купе с их угловатостью и скованностью на фоне частой экзальтированности пациентов стала причиной для названия расстройства синдромом счастливой куклы.
У них может наблюдаться как пониженный мышечный тонус туловища (гипотония), так и увеличенный мышечный тонус конечностей (гипертония), а также аномально преувеличенные рефлекторные реакции (гиперрефлексия). У некоторых детей развивается мелкий тремор рук и ног. Эти нарушения могут проявиться в возрасте от 6 месяцев до 1 года.
Освоение некоторых этапных навыков развития моторики – например, ходьбы – происходит с задержкой.
В случаях относительно легкой формы болезни, дети могут начать ходить в 2-3 года. В более тяжелых случаях прямохождение замедлено и происходит рывками – «ригидно», что тоже напоминает движения марионетки. Некоторые дети не ходят до возраста от 5 до 10 лет. Примерно 10% пациентов не способны перемещаться без посторонней помощи.
История изучения болезни
Рассматриваемая болезнь обязана своим названием врачу-педиатру Гарри Ангельману, который впервые диагностировал отклонение в 1965 году и назвал пациентов кукольными детьми.
Все три пациента имели разную симптоматику, и изначально сформировалось мнение о том, что заболевания у всех разные. Но в итоге врач обнаружил общие признаки и поставленный в итоге диагноз базировался только на клинических исследованиях, а не на научных доказательствах, поскольку сложные генетические исследования были недоступны лабораторному оборудованию того времени. Именно поэтому Ангельман не сразу опубликовал свои размышления в медицинском журнале.
Именно картина «Мальчик-марионетка» подарила специалисту уверенность в собственной правоте. Картина отображает смеющегося мальчика, который очень напомнил Гарри его пациентов и он решает написать о них общую статью, которая в дальнейшем получила название «Дети-марионетки».
Признаки синдрома Ангельмана
В 1965 году издается научная работа, интерес о которой был забыт до восьмидесятых, когда упоминания о патологии появились в медицинской среде. Закономерность отсутствия части 15-й хромосомы была установлена только в 1987 году. Поскольку диагноз «синдром счастливой марионетки» унижал и пугал многих родителей, было решено использовать фамилию Ангельмана. Среди современных ученых, изучающих данное явление, стоит выделить М. Б. Миронова и К. Ю. Мухина.
Синдром Ангельмана диагностика
Это заболевание диагностируется после генетического анализа пятнадцатой хромосомы. Диагностику назначают новорожденным с гипотонусом (пониженным мышечным тонусом), при отставании в развитии речи, а также общей моторики
Родителям следует обратить внимание на специфическое выражение лица, мелкий тремор, хаотические, а также порывистые движения конечностей, негнущиеся ноги во время походки и очень частый смех
Методы анализа включают флуоресцентную гибридизацию in situ, метилирование ДНК области 15q11- q13, а также анализ прямой мутации в гене UBE3A и анализ мутации импринтингового центра.
Существует отдельная группа людей, имеющая все признаки синдрома Ангельмана, однако при этом результаты всех вышеописанных анализов остаются в норме.
Прогноз
Прогноз при синдроме Ангельмана зависит от характера хромосомной аномалии и своевременности ее обнаружения. Тяжелее всего приходится тем деткам, чья 15 хромосома содержит «пропуски» генов (делеция). Вероятность ходить и разговаривать у таких пациентов чрезвычайно мала. Остальные случаи при внимательном подходе и любви к своему ребенку поддаются коррекции.
Стать полноценными членами общества такие больные, увы, не смогут, при всем том, что они далеко не глупы, понимают речь и ее смысл. Вот только проблемы с общением у них остаются на всю жизнь. Пациентов с детства можно обучить языку жестов, но нельзя заставить общаться при помощи слов. Лексикон «говорящих» больных ограничивается минимумом слов, употребляемых в быту (5-15 слов).
Что касается продолжительности жизни и общего состояния здоровья больных с синдромом Ангельмана, то здесь цифры колеблются на средних показателях. Во взрослой жизни пациенты в основном сталкиваются с такими проблемами со здоровьем, как сколиоз и ожирение, которые при правильном подходе к лечению не опасны для жизни.
Лечение и профилактика
На данный момент специфического лечения синдрома Ангельмана не существует.
Тем не менее, как только диагностируется заболевание, необходим надлежащий уход. Оптимальный уход должен быть как можно ранним, глобальным и подстроенным под отдельно взятого ребенка.
Глобально можно выделить следующие области:
- Физиотерапия и психомоторная реабилитация: чтобы способствовать приобретению навыков ходьбы и изучению повседневных жестов.
- Создание программы для развития альтернативной коммуникации с использованием знаков, изображений или пиктограмм позволяет добиться значительного прогресса для ребенка. Также возможно создать умственные упражнения, которые способствуют общению со взрослыми или с другими детьми.
- Нарушения сна требуют поведенческого лечения (использование ритуалов сна) или медикаментов.
- Противоэпилептические процедуры необходимы в случае судорог и назначаются чаще всего в долгосрочной перспективе.
- Миоклонус можно улучшить с помощью специального лечения.